CARDIAC + MYOPATHY
 | |
| Also see: Selective disorders of cardiac muscle |
Carnitine Disorders
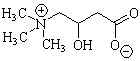
- Carnitine metabolism: General principles
- Carnitine: General
- Sources
- Dietary (75%): Meat, fish, dairy products
- Synthesis: Endogenous
- From lysine & methionine in liver, brain & kidney
- Synthetic pathway not present in muscle or heart
- Sources
- Distribution: 90% in muscle
- Low molecular weight polar compound
- Types
- Active form: L-carnitine
- Free carnitine (or L-carnitine): Nonacetylated form
- Acyl-L-carnitine
- All short-, medium- & long-chain esters
- Involved in transfer of acyl groups from acyl coenzyme A (CoA)
- Carnitine: General
- Turnover
- Absorption: Transport-mediated from GI tract
- Reabsorption
- Saturable renal tubular
- 95–99% of filtered load
- Vegetarians excrete markedly less L-carnitine than omnivores
- Preferential excretion of short-chain carnitine esters
- Plays roles in fatty acid metabolism: Oxidation of long-chain fatty acids
- Transport of fatty acids from cytoplasm to mitochondria
- Conversion of fatty acids to Fatty acid-CoA
- Carnitine & Carnitine palmitoyltransferases I & II mediate transport
- Transport of fatty acids from cytoplasm to mitochondria
- Modulates cellular & mitochondrial ratio of acyl CoA to free CoA
- Transfer of acetyl and other short-chain acyl groups from peroxisomes to mitochondria
- Oxidation of branched chain amino acids
- Reesterification of triacylglycerol in the endoplasmic reticulum before secretion as very low density lipoproteins
- Stabilization of cell membranes by removing long-chain acyl CoAs
- Removal of excess acyl groups from the body
- Mitochondrial oxidation of fatty acids provides energy source
- Chief energy sources for: Prolonged fasting; Skeletal muscle during exercise; Cardiac muscle
- Primary: Due to deficient transport of carnitine into cells
- Secondary
- Free carnitine ® acyl-carnitine esters ® Lost in urine
- Organic acidosis: Similar metabolic profile to Reyes & Valproate hepatic encephalopathy
- Renal disease: End stage; Dialysis
- Long term dialysis (Especially > 12 months): Reduces skeltal muscle carnitine by 30% to 50%
- Reduced Buffering of toxic acyl-CoA esters
- Inhibition of mitochondrial systems
- Coma after a period of starvation
- Hypoketosis: Low serum ketone concentrations
- Cardiomyopathy
- Muscle: Weakness
- LCAD

- MCAD
- Plasma-membrane carnitine transporter
- Carnitine palmitoyltransferase I (CPT I)
- CPT II deficiency
- SCAD deficiency : 2 phenotypes
● ? Autosomal Recessive
- Clinical
- Onset age: Childhood - Early adult
- Weakness: Symmetric; Proximal; ± Face & Tongue
- No pain or rhabdomyolysis
- Progression: Usually slow; Rarely acute; ? Exacerbations 2° pregnancy
- Cardiac: Cardiomyopathy & Congestive heart failure in some patients
- Serum CK: Moderately high
- EMG: Myopathic
- Muscle biopsy
- Lipid storage
- Low carnitine levels
- Diet: Low fat
- Carnitine: 2 to 4 g/day (Children 100 mg/kg/day)
- Drugs: ? Prednisone; Riboflavin
● Na+-dependent carnitine transporter (OCTN2; SLC22A5)
- Genetics: Mutations
- Most produce null alleles
- Homozygous missense (E452K): Disease onset 7 years; Cardiomyopathy
- Mutation effects on carnitine: Reduced carnitine-transporter
- Impaired muscle uptake
- Decreased renal tubular reabsorption
- OCTN2 protein
- Family: Carnitine/organic cation transporter
- Expressed on surface membrane of cardiomyocytes
- Upregulated by: Nuclear receptor PPARα
- Other substrates: Quinidine; Verapamil
- Function: Transports carnitine across cell membranes
- Onset age: Infancy to 1st decade; Intrafamilial variation
- General: Fatigability; Vomiting; Abdominal pain; Low height & weight
- Hypoglycemia: May occur in infants
- Encephalopathy: Episodic; r/o Reyes syndrome
- Myopathy: Generalized weakness
- Cardiomyopathy
- Dilated
- Ventricular hypertrophy
- Heterozygous OCTN2 mutations: Predisposed to late-onset benign cardiac hypertrophy
- Cardiac failure may occur < 10 years
- Carnitine levels (Total & Free)
- Low or absent in plasma & many tissues
- Total carnitine in muscle: < 5%
- Hyperammonemia
- Urine: Low dicarboxylic acids; Leakage of carnitine
- Normal in 2° carnitine deficiencies
- Lipid storage (Droplets): Predominantly in type 1 muscle fibers
- Type 2 muscle fiber atrophy
- Acute episode: Intravenous glucose infusion
- Avoid fasting
- Low fat diet
- L-carnitine (100 - 150 mg/kg/d oral)
- Pathogenesis
- Reduced Protein synthesis ® Reduced number of Carnitine carriers
- Competitive inhibition for carnitine uptake
- Increased excretion
- Hypotension: Intradialytic
- Heart failure
- Anemia:Eerythropoietin-resistant anemia
- Muscle: Weakness; Low exercise capacity
- Systemic disease
- Malnutrition
- Sepsis
- Organ failure: Renal; Liver; Endocrine
- Organic aciduria
- Reye's syndrome
- Chronic myopathies
● CPT2
- General
- Genetics 15
- > 70 different mutations identified
- Null mutations produce: Lethal neonatal
- Amino acid substitution (Missense) mutations: Produce varyiung severity depending on degree of effect on enzyme activity
- R631C
- Present in both infantile & adult onset CPT2 deficiency syndromes
- Associated with Calabria
- Mutations
- Some mutations produce thermolabile protein
- External database
- Genetics 15
- Biochemistry: Suggests CPT2- or carnitine-acylcarnitine-translocase- deficiency
- Elevations of long-chain acylcarnitines: C14:0-, C16:0-, C16:1- and C18:1-acylcarnitines
- Increased ratio of (C16 + C18:1)/C2
- May be normal in anabolic conditions
- See: Acylcarnitine patterns
- Activity of CPT II: Represents 20% to 40% of total CPT activity
- Disease
- Muscle symptoms: Enzyme activity < 25%
- Severe muscle phenotype
- Enzyme activity < 15%
- Homozygous mutations: S113L (15%) or R631C (7%)
- Heterzygous with 1 Null
- Adult onset, Myopathy
- Infant onset
- Lethal neonatal
- Acute infection-induced encephalopathy, Susceptibility 4 (IIAE4)
- Epidemiology: Most common metabolic cause of repeated myoglobinuria
- Genetics
- Clinical syndromes & Mutations
- ~20 mutations described
- Most cases with common mutations ± Heterozygous for 2nd mutation
- Ser113Leu: 60% of adult myopathy cases; Mild disease
- 413delAG: Ashkenazi Jews
- Pro50His: 10%
- Clinical syndromes & Mutations
- Arg503Cys: Malignant hyperthermia with variable myopathy
- May contribute to reduced CPT2 activity in combination with disease allele
- Single isoform
- Homotetramer
- Function
- Mediates transport of Fatty acid-CoA across inner mitochondrial membrane
- Involved in fatty acid β-oxidation
- Metabolic defect promotes glycogen depletion
- Onset age
- Adolescent or Adult
- Mean 13 years
- Range 1 to 40 years
- No relation between early onset age & genotype
- Triggers: Activities requiring fatty acid oxidation
- Prolonged exercise
- Cold
- Diet: Low-carbohydrate, high-fat diet; Fasting
- Infections
- Valproate treatment 7
- Anaesthesia
- Early: Normal strength between attacks
- Late: Weakness in some patients
- Myalgias: Less severe than VLCAD deficiency
- Cramps
- Serum CK
- Normal or mildly elevated (50%) between episodes
- High with rhabdomyolysis
- High ratio of: (Palmitoylcarnitine (C16:0) + Oleoylcarnitine (C18:1))/Acetylcarnitine (C2)
- Normal serum carnitine
- Morphology: Normal or Varied fiber size (Small type 1)
- Type 2 muscle fiber predominence
- Lipid: Increased in muscle fibers (50%)
- CPT activity: Reduced by 80% to 90% in homozygotes
- Exercise with fasting or infection
- Dehydration
- Excess heat exposure
- Genetics
- Most cases with different mutations
- Residual enzyme activity: Minimal (< 10%)
- Tyr628Ser, Glu174Lys, Phe383Tyr mutations
- Severe clinical disease: Infant form
- Lower CPT2 enzyme activity
- Hepatic involvement
- Most cases with different mutations
- Hepatic
- Cardiac
- Muscle
- Genetics
- Homozygosity or compound heterozygosity for any null mutations
- Clinical
- Cardiac abnormalities: Arrythmias
- Congenital anomalies
- Neurologic: Lethargy, Seizures, Hypotonia, Hyperreflexia
- Death: 1st weeks
- Hypoketotic hypoglycemia
- Carnitine: Reduced total & free
- CPT II activity: Severely reduced
- Epidemiology: Chinese &. Japanese patients
- Genetics: Missense mutations, Phe352Cys & Val368Ile
- CPT2 protein: Mutation causes thermolability
- Clinical
- Fever
- Seizures
- Coma
- Multiorgan failure
- Brain edema
- Laboratory
- Acylcarnitine ratios: High
● SLC25A20 (CACT)
- Protein
- Mitochondrial-membrane carrier protein
- Shuttles substrates between cytosol & intramitochondrial matrix space
- Clinical
- Encephalopathy: Fasting induced coma & seizures
- Cardiomyopathy
- Muscle weakness
- Respiratory: Episodic neonatal apnea
- Course: High neonatal mortality
- Hypoketosis
- Hypoglycemia
- Hyperammonemia
- Peritoneal dialysis with a permanent Tenckof catheter in situ
- Enteral feeding
- Frequent
- High calories
- Low protein
- Long-chain fatty acids
- Medium-chain triglyceride oil
● Electron transfer flavoprotein, α polypeptide (ETFA)
- Allelic with: Coenzyme Q10 deficiency
- Clinical
- Onset age
- Usual: Adult; 3rd & 4th decade
- Range: 6 to 64 years
- Myopathy 2
- Proximal
- Weakness: May become severe
- Myalgias: Some patients
- Progression: Subacute over months
- Onset age
- Urine: High glutaric & ethylmalonic acids
- Consistent with defective dehydrogenation of isovaleryl CoA & butyryl CoA
- Serum CK: High; 2x to 20x
- EMG: Myopathic, Neuropathic or Normal
- Muscle biochemistry
- Carnitine levels Reduced
- Catalytic activity of mitochondrial complex I Reduced
- Lipid storage: Type 1 fibers
- ? Vacuolar myopathy
- SDH stain: Reduced intensity
- Riboflavin (100 mg/day)
- ± Carnitine (4 g/day)
- Coenzyme Q10
- Corticosteroids
- Acidosis
- Hypoglycemia
- Sweaty feet odor
- Death
- Onset: Infantile
- Respiratory insufficiency
- Chronic central alveolar hypoventilation
- Nocturnal respiratory disorder (Ondine syndrome): Sleep apnea
- Acidosis
- Low carnitine
- Lipid storage myopathy
- ? Disorder of sarcolemmal carnitine carriers
● Short-chain acyl-CoA dehydrogenase (ACADS)
- Infant onset
- Clinical
- Failure to thrive
- Developmental delay
- Seizures
- Hypotonia
- Muscle weakness
- Clinical
- Laboratory
- Acute metabolic acidosis
- Ethylmalonate excretion
- SCAD deficiency: Generalized
- No episodes of nonketotic hypoglycemia
- Clinical
- Ophthalmoplegia
- Ptosis
- Weakness
- Scoliosis
- Laboratory
- Muscle
- Lipid storage
- Multicores
- SCAD deficiency: Localized to skeletal muscle
- Muscle
Dilated cardiomyopathies ± Myopathy
| Myofibrillar myopathy (ARVC) Barth syndrome: Tafazzins; Xq28 Barth-like syndrome: mtRNA Leu Dilated cardiomyopathy (Isolated): 1q32; 9q13; 10q22 Dilated Cardiomyopathy with Ataxia: DNAJC19; 3q26 Dystrophinopathies: Xp21 Familial with Conduction Defect& Muscular dystrophy (CMD 1F): 6q23 Familial with conduction defect without dystrophy CMD 1A: Lamin A/C; 1p11-q11 CMD 1E: 3p25-p22 McLeod syndrome: XK protein; Xp21 Mitochondrial Myopathy + Cardiomyopathy: DPM3; 1q12 Nemaline (Rod) myopathy Other familial dilated cardiomyopathy without myopathy Selenium deficiency Also see: Dilated cardiomyopathy without myopathy |
McLeod syndrome8
● Kell group protein (XK membrane transport protein)
- Genetics
- Mutations
- Usually produce stop codons
- Types
- Frameshifts with deletions
- Point mutations: Stop codons
- Missense mutations: 2 described
- Splice site
- Evenly distributed through gene
- No clear correlation between mutation & phenotype
- Mutations
- Duchenne dystrophy
- Chronic granulomatous disease
- Retinitis pigmentosa
- Location: Erythrocyte membrane
- Function
- Zinc metalloproteinase
- Cleaves endothelin-3 to bioactive peptide
- Homology to CED-8: ? Role in programmed cell death
- McLeod syndrome: Reduced expression on erythrocytes
- ? Associated proteins
- Huntingtin: Huntington's disease
- Chorein: Autosomal-recessive Chorea-Acanthocytosis
- Muscle location
- Type II muscle fibers
- Intracellular: Sarcoplasmic reticulum
- Staining is absent in McLeod myopathy
- Disease frequency: 0.5 to 1 per 100,000
- Onset
- Age: Adult; Range 27 to 72; Mean 5th decade
- Signs: Fatigue; Movement disorder; Seizure
- Weakness (65%)
- Severity: Usually mild; Occasionally severe or none
- Distribution: Proximal; Symmetric; Legs > Arms
- Increases with age
- Other muscle features
- Muscle atrophy: May be generalized
- Fatigue with exercise
- Rhabdomyolysis9: Associated with hyperkinesis & neuroleptic drugs
- Serum CK: High
- Sensory loss: Reduced vibration sense in feet (40%)
- Tendon reflexes: Reduced; Legs (90%) ± Arms (60%)
- Symptoms: Rare
- Movement disorders
- Onset: 2nd to 6th decade
- Distribution: Limbs; Trunk; Face
- Types: Chorea (100%); Dystonia; Face hyperkinesia; Vocalizations; Dysarthria
- Hepato-Splenomegaly (40%)
- Cardiomyopathy (66%)
- Onset > 40 years
- Cardiomegaly: Dilated or restrictive
- Atrial fibrillation
- Congestive heart failure
- Often cause of death
- Hematologic
- Acanthocytosis
- Weak espression of Kell red blood cell antigens
- Compensated hemolysis
- Chronic granulomatous disease
- Results from a contiguous gene deletion
- Autologous blood donation: Avoids incompatibility hazards
- Symptomatic treatment: Epilepsy, Cardiac & Psychiatric features
- Chorea: Porly responsive to treatment
- Acanthocytosis: Mild
- CNS: Chorea; Occasional psychiatric syndrome
- Serum CK: Mildly increased
- Hematologic
- Acanthocytosis6: 100%
- Thorny shaped red cells
- May be marked: 35%
- Differential diagnosis: A-β-lipoproteinemia; Chorea-Acanthocytosis
- Acanthocytosis6: 100%
- CK: High (100%); 2,000 to 5,000; Range 1.5 to 15x normal
- LDH: High (91%)
- Myopathic (14%)
- Neuropathic (79%)
- CMAPs: Small amplitude
- SNAPs: Small amplitude
- Conduction velocities: Normal
- Variation in muscle fiber size
- Nuclei: Internal
- Necrosis & Regeneration of muscle fibers
- Occasional
- Severe cases: May be prominent
- Cell infiltrates: Occasional patients; Predominantly macrophages; Few lymphocytes
- Fiber type changes
- Type I predominance
- Type II atrophy
- Axonal loss
- ± Demyelination
- MRI: Sparing of quadriceps
- CT: Asymmetric involvement of thighs & lower legs (calves)
- Caudate atrophy
- MR: Abnormal signal in basal ganglia, increased T2 in lateral putamen
X-linked dilated cardiomyopathy (Barth syndrome)
● Tafazzin (G4.5 gene; TAZ)
- Genetics
- Mutations
- Locations: In all exons & some adjacent intron sequences
- Stop & Missense
- No strong genotype-phenotype correlations
- ? More severe heart disease with some exon 8 mutations
- Allelic with Cardiomyopathy, Dilated 3A (X-linked fatal infantile)
- Also see: Barth-like syndrome with mtRNA Leu mutations
- Mutations
- Superfamily: Acyl transferases involved in phospholipid synthesis
- Isoforms: 10 different proteins
- Membrane anchored types
- Contain hydrophobic N-terminus (1st 2 exons)
- High levels in heart & skeletal muscle
- Membrane anchored types
- Cytosolic types
- Lack hydrophobic sequence
- High levels in leukocytes & fibroblasts
- Phospholipid acyltransferases
- Remodeling of cardiolipin
- Cardiolipin (CL)
- Mitochondrial-specific phospholipid
- Major component of inner mitochondrial membrane
- Tetralinoleoyl-cardiolipin (L4-CL): Dominant species of cardiolipin in heart & skeletal muscle
- Barth changes
- Tetralinoleoyl-cardiolipin reduced
- Membrane destabilization: Generalized electron transport chain dysfunction
 From: The Barth Syndrome Foundation |
- Onset
- Infancy
- Hypotonia
- Cardiomyopathy: Neonatal to < 1 year
- Low birth weight
- Myopathy: Non-progressive; Relatively mild
- Early
- Hypotonia
- Delayed motor development
- Weakness
- Proximal > Distal: May be diffuse
- Waddling gait
- Gower's sign
- Mild: No respiratory insufficiency or wheelchair dependence
- Myopathic facies: Some patients
- Muscle mass: Reduced
- Fatigue
- Myalgias
- Early
- Learning disability
- Delays: Mathematics; Visual special tasks & short-term memory
- Headaches
- Proportionate (-2 SD)
- May eventually grow to normal height
- Delayed bone age
- Full cheeks
- Prominent ears
- Deep-set eyes
- Noncompaction of left ventricular myocardium
- Isolated: No additional cardiac defects (pulmonary atresia)
- Lethal early course
- Ultrasonogram: Spongy structure of left ventricular wall
- Also caused by α-dystrobrevin mutations
- Some progressive: May need cardiac transplantation
- Others resolve with time
- Serum CK: Normal
- Muscle: Mild myopathic
- ± Lipid accumulation, or Type 1 fiber predominance
- Mitochondrial disorder
- Electron transfer chain variably affected: Complex I deficiency + other
- Morphologically abnormal inner mitochondrial membranes in cardiac muscle
- Deficient mitochondrial phospholipid: Tetralinoleoyl-Cardiolipin (L4-CL)
- Endomyocardium: Mitochondria with tightly packed, concentric cristae
- Blood: Increased 3-methylglutaconic; Low cholesterol
- Immune deficiency
- Neutropenia: Recurrent
- Neutrophils 0 to 500
- Maturation stop at promyelocyte stage
- May be normal when patient is well
- May predispose to lethal infections in neonatal period
- Associated with chronic aphthous stomatitis: Usually due to Candida
- Treatment: ? Granulocyte colony stimulating factor (G-CSF)
- Neutropenia: Recurrent
- Increased levels of some branched-chain organic acids
- 3-methylglutaconic acid
- 3-methylglutaric acid
- 2-ethylhydracrylic acid
- 3-methylglutaconic acid may be high normal in urine in older patients
- Some: Death in childhood due to cardiac failure or sepsis
- Many: Slow improvement in cardiac, motor & infectious signs
- Clinically normal
- Skewed X inactivation common
- X chromosome with mutation often selectively inactivated
- Usually occurs when mutated gene is on maternal chromosome
- Inactivation may be > 95%
- Other skewed inactivation syndromes
- Wiskott-Aldrich syndrome
- α-thalassemia/mental retardation syndrome
- Bruton X-linked agammaglobulinemia
- Increases in older females
Barth-like syndrome with mitochondrial mtRNA Leu mutation5
● Mitochondrial tRNA Leu (MTTL1)
- Genetics
- Maternal inheritance
- Mutation: A3243G
- Onset: 1st months
- Failure to thrive
- Motor: Weakness; Milestones severely delayed; Respiratory failure
- Cardiomyopathy: Dilated; Progress to congestive heart failure
- Lactic acidosis
- CBC normal
- Serum CK: Mildly elevated, early
- Serum carnitine: Mildly reduced
- Urine: High 3-methylglutaconic & 2-ethylhydracrylic acid
- Cholesterol: Low in blood
- Muscle
- Histochemistry
- Type 2 predominance
- Varied fiber size
- Ragged red fibers; Diffusely reduced COX activity; SDH increased
- Mitochondrial biochemistry: Low Complexes I & IV
- Histochemistry
Dilated Cardiomyopathy with Ataxia (DCMA)11
● DNAJC19 (TIM14)
- Epidemiology: Canadian Dariusleut Hutterite population
- Mutation: Splicing site; IVS3-1 G>C
- DNAJC19 protein
- Location: Mitochondrial inner membrane protein (TIM)
- ? Component of TIM23 inner membrane translocase complex
- Structure: DNAJ domain-containing
- Function: ? Involved in molecular chaperone systems of Hsp70/Hsp40 type
- Other TIMM protein disorder: Mohr-Tranebjaerg syndrome
- Location: Mitochondrial inner membrane protein (TIM)
- Cardiomyopathy: Dilated
- Conduction defects: Long Q-T syndrome
- Early-onset: < 3 years
- Ataxia
- Motor delay
- Course
- 100% after 2 years of age
- Non-progressive
- Independent ambulation achieved
- Optic atrophy: Some patients
- Mental retardation: Mild
- Seizures: Occasional patient
- Testicular dysgenesis: Cryptorchidism to severe perineal hypospadias
- Growth failure (100%)
- Organic aciduria (Methylglutaconic aciduria type V)
- 3-methylglutaconic
- 3-methylglutaric
- Other organic aciduria: Barth syndrome
- Hepatic enzymes: Mildly elevated
- Anemia: Normochromic, Microcytic
Isolated dilated cardiomyopathy
● Chromosome 1q32
● Chromosome 9q13
● Chromosome 10q22-q24
Hypertrophic cardiomyopathy with myopathy
- Hypertrophic cardiomyopathy with central cores in skeletal muscle
● Myosin - Cardiac β heavy chain (MYH7); Chromosome 14q11.2; Dominant - Allelic disorders
- Clinical features
- Cardiomyopathy: Hypertrophic
- Strength: Normal
- Muscle biopsy
- Central cores
- Type I muscle fiber predominance
● Myosin light chain, Ventricular and skeletal slow type (MYL3); Chromosome 3p21.31; Dominant
l ? Mutations at hinge region between heavy & light myosin chains
- Muscle biopsy: Myopathic with ragged red fibers
- Other: Familial hypertrophic cardiomyopathy 8
Mitochondrial
Cardiomyopathy: Hypertrophic or Dilated
- Selective Cardiomyopathy
- Congenital & infantile
- Kearns-Sayre
- Multisystem
- PEO
- Congenital muscular dystrophy with mitochondrial structural change
Other disorders with cardiomyopathy
- Amyloid
- Congenital myopathy with spindle excess
- Desmin (Cardiomyopathy) Myopathy
- Encephalopathy with necrotizing myopathy, cardiomyopathy & cataracts
● Autosomal recessive - Limb Girdle Dystrophy
- Dysrhythmias
- Type 2D: Rare
Drugs + Cardiomyopathy
|
Isolated cardiomyopathies; Hereditary
- Hypertrophic cardiomyopathy
- Dilated cardiomyopathy
- Errors of Fatty acid oxidation - 2° disorders of carnitine metabolism
(See 1° disorders of Carnitine metabolism) - Mitochondrial
- Other
- Congestive cardiomyopathy with conduction defects
- Amyloid
MULIBREY NANISM
● Tripartite motif-containing protein 37 (TRIM37) - Mutations
- Frameshift, deletion or insertion
- Produce truncated protein
- Disease category: Peroxisomal biogenesis disorder
- Protein
- Zinc finger protein: RING-B-box-coiled-coil (RBCC) family
- Contains TRAF domain: Interacts with other proteins
- Coiled-coil domain: Homo-oligomerization; Subcellular localization
- Most common in Finland
- Onset: Birth (Prenatal)
- Muscle: Hypotonia 80%
- Cardiac: Constrictive pericarditis
- Skeletal
- Prenatal growth failure
- Slender stature
- Cystic dysplasia of tibia (30%)
- Long shallow (J-shaped) sella turcica (95%)
- GI: Hepatomegaly
- Skin: Naevi flammei (70%)
- Small voice
- Eye
- Choroid hypoplasia
- Retinal yellowish dots & pigment dispersion
- Wilms tumor (4%)
- Endocrine: Gland hypoplasia
- CNS: Probably normal
Return to Myopathy& NMJ Index
References
1. Neuromuscular Disorders 1999;9:320-322
2. Brain 1999;122:2401-2411
3. JNNP 2000;69:655-657
4. J Pediatr 1999;135:273-276; American Journal of Medical Genetics 2004;126A:349–354
5. Am J Med Genet 2001;99:83-93
6. J Neurol 2001;248:87-94
7. Neuromuscular Disorders 2001;11:757-759
8. Ann Neurol 2001;November On-Line
9. Muscle Nerve 2002;June On-Line
10. Neurology 2002;59:1046-1051
11. J Med Genet 2005 Aug 3
12. Neurology 2008;71:260–264
13. Semin Dial 2006;19:323-328
14. New Eng J Med 2009;360:838-840
15. Molecular Genetics and Metabolism 2008;94:422–427
7/2/2014
No comments:
Post a Comment